
Perhaps the most perplexing aspect of myalgic encephalomyelitis is not any feature of the disease which, after all, has been subject to limited investigation, but the deafening non-response of the world’s governments, the American and British governments especially, which is a response against human decency.
Nevertheless, in spite of this, today there are several groups producing high-quality research and there are reasons to be cautiously optimistic that if these groups can be funded at decent levels,* the mechanism which causes disease, and also a treatment, can be found.
Here are two such recent studies:
*As opposed to the shoestring budgets of today.
Peroxisome abnormalities found in a study by Xiaoyu Che, Christopher Brydges, Ian Lipkin and Colleagues
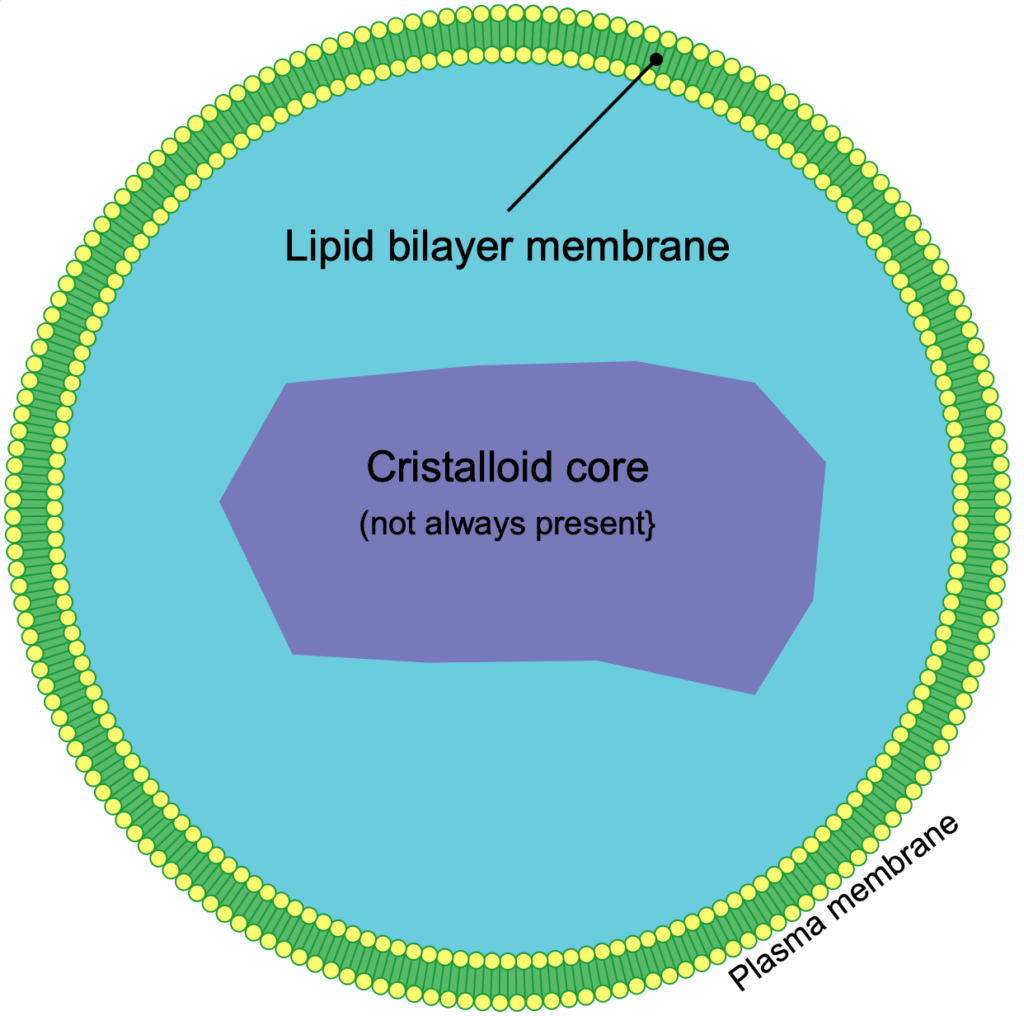
Plasma levels of plasmalogens are decreased in patients with myalgic encephalomyelitis/chronic fatigue syndrome suggesting peroxisome dysfunction.
– Che, Brydges, Lipkin, et al. [1]
7/19 Update: this article has now been published [2]; Read the fulltext here.
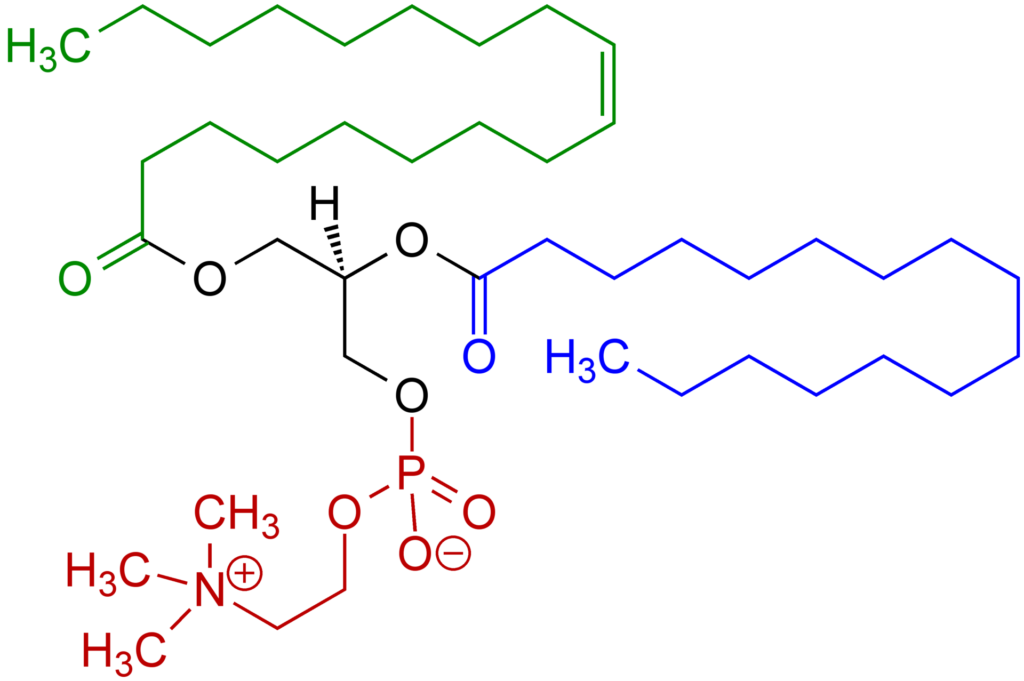
Peroxisomes are small organelles found in virtually all eukaryotic cells [3]. They are most concerned with oxidative reactions including the catabolism of long-chain fatty acids, branched chain fatty acids, bile acids and polyamides and the biosynthesis of plasmalogens (fats which based on a glycerol backbone with an ester linked lipid at the second carbon) [4]. Their name comes from their role in producing and scavenging Hydrogen Peroxide, a potent oxidizing molecule. Another connection to metabolism is that peroxisomes contain 10% of the activity of two enzymes involved in the Pentose Phosphate Pathway [5] which is necessary for the synthesis of nucleotides. More approachably, the film Lorenzo’s Oil is about two parents’ search for treatment for their son with an inherited peroxisomal disorder where the loss of a transporter protein leads to a buildup of very long chain fatty acids in the body [6].

This study by Xiaoyu Che, Christopher Brydges, Ian Lipkin and others investigated 888 metabolites in over 100 patients and 90 controls [1]. Many of the researchers (such as Komaroff, Batemen, Hornig, Montoya, Levine, Vernon and Peterson) will be very familiar to most patients. The results used a variety of statistical analysis and found, most prominently, decreased levels of plasmalogens.
Many other lipid metabolites were also decreased

Additionally to decreased plasmalogens, the study found patients had reduced levels of sphingomyelins, unsaturated phospholipid ethers, unsaturated ceramides, carnitines, saturated lysophospholipids, unsaturated lysophosphatidylethanolamines, prostaglandins and saturated triglycerides [1]. Patients also had increased levels of hydroxy-eicosapentaenoic acid, dicarboxylic acids and long-chain unsaturated triglycerides [1]. The study also found metabolites which correlated with physical symptoms such as fatigue (interestingly, levels of long chain unsaturated triglycerides were positively correlated with fatigue in male patients.) [1].
It is striking how many of these decreased metabolites either belong to the lipid group (including the plasmalogens, phosphatidylcholine and products of phosphatidylcholine) or are involved in the metabolism of lipids! Carnitine, which was also reduced, is involved in the transport of long-chain fatty acids into the mitochondria [7].
What does this all mean? For one thing, since plasmalogens are protective against oxidative stress [8, 9] decreased plasmalogens levels could impair the membrane function of membranes (including the peroxisomes) and lead to more susceptibility to oxidative stress [1]. If I follow correctly, the authors (the section I am referring to begins: “We posit that this crosstalk”) assume that diminished membrane integrity of peroxisomes in ME could lead to diminished oxidation of long-chain fatty acids since the mitochondria is dependent on the peroxisome for breaking down these into short-chain fatty acids the mitochondria can oxidize [1, 10].
The decreased levels of carnitine (a molecule involved in transporting fatty acids into the mitochondria) also found are another thing that could contribute to mitochondrial dysfunction and toxic levels of long-chain fatty acids [1].
{kind=link}
{kind=link}
Thirdly the authors found a decrease in phosphatidylcholines (PD) and downstream products of PDs (ceramides, sphingomyelins, lysophosphatidylcholines, phospholipid ethers, prostaglandin D2 (PGD2) and prostaglandin F2α (PGF2α)) in myalgic encephalomyelitis patients. As with the plasmalogens, PDs and many of their derivatives play important roles in lipid membrane structure and cell signaling cascades. Loss of PDs in patients’ cells could affect the mitochondria translocases [1] and lead to a loss in energy product. The authors further suggest that deficiencies in these molecules could lead to membrane dysfunction especially of membrane-bound receptors like G-protein coupled receptors [1].
Choline was also found to be reduced but the result did not reach statistical significance [1]. In addition to the important role in membrane structure already mentioned, choline is used to provide a methyl group to make epinephrine [1]–potentially tying the lipid abnormalities to the sympathetic nervous system (the “fight or flight” side of the nervous system).
Abnormal products of the TCA Cycle
Two intermediates of the TCA-cycle (also known as the Krebs Cycle and Citric Acid Cycle), α-ketoglutarate and succinate, were found to be elevated in patients [1]. The main product of this cycle is NADH which is used to produce ATP. It is notable that alpha-ketoglutarate and succinate occur next to each other in the TCA cycle, perhaps suggesting that there is a block somewhere downstream, or that some other process is dumping into the cycle at this point in myalgic encephalomyelitis. Alpha-ketoglutarate is both also used to make glutamate-an important neurotransmitter (Older patients may even remember Paul Cheney, a famous physician who was involved with the Incline Village Outbreak, postulated there is a glutamate excitotoxicity in ME) and also a product of glutamate deamination [11] .
Unfortunately excesses of TCA cycle intermediates is actually a bad thing. The authors note that excess alpha-ketoglutarate is associated with impairments in pyruvate metabolism and even cell death [12]. Excess succinate has been associated with increased oxidative stress, neuronal degeneration and stabilization of the transcription factor hypoxia inducible factor 1-alpha (HIF 1a) [13, 12].
In Summation: an important study which points to the junction of peroxisomes and mitochondria.
Much of the molecules which were abnormal were molecules associated with the peroxisome (such as plasmogenons) or with the mitochondria (such as carnitine and increased long-chain fatty acids). The interpretation that there could be disruption at the junction between the peroxisomes and the mitochondria, with the peroxisome not creating products to feed into mitochondrial respiration makes a lot of sense. Also like the disease in Lorenzo’s Oil, Adrenoleukodystrophy, we know that, at least, inherited disorders of the peroxisomes can produce very severe phenotypes: a fact that is consistent with the severe disruption to physical and mental activity seen in ME.
Another finding which could be relevant to the claim that decreased plasmalogens lead to oxidative stress and possibly the production of toxic oxidized lipids is Ron Davis’s finding that many patients are deficient in the metal manganese [14]. Manganese is an important cofactor in glycolysis [15] and in the mitochondrial superoxide dismutase, [16] a crucial enzyme in protecting the mitochondria from oxidative stress. Assuming both results hold up, a deficiency in manganese could lead to increased susceptibility to oxidative stress and could support the idea in this paper that oxidative stress due to altered lipid metabolism is important in this disease [1].
Considering how many of the abnormally low molecules were lipids or phospholipids which play in important role in cell and organelle membrane integrity the authors suggested that membrane integrity could be compromised in ME and that this could be a cause of dysfunction in the peroxisome, the mitochondria and various signaling receptors which are embedded in the membrane [1].
Decreased Production or Increased Turnover?
It isn’t clear to me whether the low levels of these molecules, such as the plasmalogens, were due to decreased production or to increased turnover (though maybe I just missed it)–that strikes me as an important thing to know. Several studies have found increased apoptosis (programmed cell death) in ME patients’ cells [17, 18, 19] so perhaps patients’ bodies are cycling through some of these lipids at a faster rate than they can be replaced. However, it is also possible that there is firstly increased production of a toxic product (oxidized triglycerides? viral RNA?) that pushes cells en masse towards the programmed death pathway. (Could it be that the altered levels of some of these important lipid constituents of cellular membranes leads to globally increased apoptosis and the resulting cell death creates a self-reinforcing shortage?)
The increase in alpha-ketoglutarate and succinate provide further evidence that mitochondrial metabolism is abnormal in myalgic encephalomyelitis and serves to bolster the conclusion of earlier studies such as by Hoel and Hoel [20] and Naviaux [21].
If I am understanding correctly, Che et al articulate a potential mechanism which, I think(!), goes like this: decreased plasmalogens and structural phospholipids (from as yet unknown causes) → loss of peroxisome membrane integrity → loss of fatty acids to export to the mitochondria and diminished reactive oxygen species scavenging → carnitine depletion and production of increased levels of toxic oxidized triglycerides. This is all very testable and hopefully future studies will look into it.
We hypothesize that damage or stimulation of the
Yoshitsugi Hokama et al 2009 [22]
mitochondria results in the release of phospholipids,
which in turn induce production of an autoimmune
antibody (antiphospholipids).

There are earlier studies which point to fats in the circulation as markers, and possible effectors of the disease. In 2003 Yoshitsugi Hokama’s discovery, showing that a lipid in the sera of patients cross-reacted to the antibody used to detect ciguatera toxin [23] shot like a bolt of light across the sky. Later, in 2009, it was determined that the identity of the antigen was phospholipids associated with cardiolipin from the mitochondria [22]. Unfortunately, further studies did not take place. Assuming this result was correct, this leaves the question: what are these phospholipids doing at increased concentrations in the blood? A likely consequence of Che and Lipkin’s study is that there may be increased, toxic, oxidized, lipids in the blood of patients with ME, which could, in some measure, also vindicate this early study.
Hopefully the search for such lipids and their significance to the pathomechanism of ME will shortly begin…
Massimo Nunes, Amy Proal and colleagues find fibrinoid microclots in the blood of ME patients.
So it turns out we did find “something in the blood” of #ME/CFS patients: fibrinoid microclots + hyperactivated platelets (that you can see with your own eyes).
Amy Proal on Twitter. [24]
In another preprint–study that looks to be very significant, Amy Proal and colleagues found fibrinoid microclots in the blood of myalgic encephalomyelitis patients [25]–similar to an earlier result finding such clots in Long Covid patients [26].
Using platelet poor plasma Massimo Nunes, Etheresia Pretorius and colleagues found that when stimulated to clot the plasma from ME patients formed more extensive clotting networks. Also using fluorescence microscopy they found fibrinoid microclots at a burden greater than controls but, interestingly, lesser than acute Covid-19, long Covid-19 and type 2 Diabetes. Using TnT staining they determined these microclots are amyloid in nature and also that “Amyloid protein changes were markedly increased in thrombin-induced fibrin clots.”
The authors suggest these fibrinoid microclots could be both causing blood vessel inflammation (especially in the microvasculature) and serving as a mechanical blocking point to oxygen getting through to microcapillaries. The authors further state it is possible that these fibrinoid microclots could contribute to the pathology in a feed-forward fashion: that the presence of these amyloid microclots leads to inflammation which (by itself or with another factor) leads to more amyloid microclot formation in a repetitive cycle. If this is true, not only would the microclots form a immediate locus for potential treatments but disrupting their formation or removing them more quickly might ameliorate the disease.
‘To my knowledge, there are only five diseases that have a pathological low sedimentation level: Myalgic Encephalomyelitis, sickle-cell anemia, hereditary spherocytosis, hyper-gammaglobulinemia [and] hyper-fibrogenemia’
Byron Hyde, quoted in Osler’s Web [27]
The idea that the microcirculation of ME patients could be compromised in some way goes far back. In 1985 a pilot study by Dr. L.O. Simpson found a filtration rate difference between female patients’ blood as compared to healthy controls [28]. In a second study, Simpson found abnormally shaped red blood cells (most especially “cup forms”) in patients which could impair microcirculation [29]. Much more recently, Amit Saha, Ron Davis, and colleagues, found a reduced deformability in red blood cells of patients [30] a finding that is likely related to Simpson’s finding of abnormal erythrocyte forms.
The hypercoagulation hypothesis is also not new: it was first advanced by David Berg in the late 1990s [31]. However, subsequent follow-up study, by the reputable ME Research UK group, didn’t find evidence hypercoagulation [32] and thereafter the hypothesis received little attention. This study would have the effect of reviving this hypothesis and providing a plausible mechanism for how it could result in the intolerance to exertion (or visual/auditory/cognitive stimulation) that is one of the core characteristics of the disease.
If our blood is filled with clotted particules and has a tendency toward hypercoagulation it could also explain the long-mysterious low erythrocyte sedimentation rate found in this disease as most inflammatory diseases have the inverse finding of an elevated sedimentation rate. (Other explanations are also possible though: For example, it could be that our red blood cells have a higher negative potential (and thus repel each other more strongly), or that they have altered shapes, or membrane compositions which are resistant to stacking.)
Dr Beate Jaeger and HELP Apheresis
Dr Jaeger was also applying a treatment with which she was already familiar, namely HELP apheresis. This treatment has been used since the 1980s to remove excessive levels of lipid from the blood of patients with coronary artery disease. Dr Jaeger therefore decided that this could be adapted to remove microclots from the circulation of Long Covid patients, by a sophisticated process of filtration. The technique involves continuous removal of blood from one arm vein whence it is filtered, before being returned to the opposite arm.
–Dr. William Weir.
If Nunes, Proal, Pretorius and colleagues are correct and the higher level of microclots in myalgic encephalomyelitis is confirmed and the microclots are not merely an unpleasant side-effect of inflammation but “the thing itself” the question naturally arises: how can they be removed and would their removal effect a clinical improvement?
The following is my synopsis of a summary provided by Dr. William Weir, and shared by Jenny Wilson, of a visit by Weir to Dr. Beate Jaeger at the Mulheim Clinic in Germany.

Dr. Beate Jaeger is a German doctor who began using HELP Apheresis (a method of separating a harmful constituent in the blood from the rest of the blood that is returned to the patient; HELP is an acronym for : heparin-induced extracorporeal LDL precipitation) to remove microclots from patients with Long-Covid19 and began treating ME patients with this same procedure upon receiving results that they also had increased levels in microclots in the blood. When this material was by a South African team (Dr Pretorius’s group) they found the filtered material was composed of circulating cellular debris, clumps of fibrin, platelets and red blood cells. Dr. Jaeger believes there are also subsets where it is not microclots but lipid globules (which, if true, would seem to accord better with the Che/Lipkin study). The number of procedures needed to completely filter this debris from the blood was 2-10. Clinical improvement in Long-Covid19 was substantial and there was also improvement in the few ME patients treated thus far.
The summary concludes with these encouraging words:
The acid test of Dr Jaeger’s work will be to demonstrate not only subjective symptomatic improvement but also scientifically objective measures of improvement. Sophisticated scanning techniques as well as the recovery of fingerprint patterns will help prove the effectiveness of her treatment.
I am cautiously optimistic that, when Dr Jaeger’s results appear in the medical literature, HELP apheresis will then be adopted universally as standard treatment for ME/CFS and Long Covid. Time will tell and fingers crossed!
–Dr. William Weir
If the finding of microclots and other debris in ME patients’ blood bears out, aside from all else that can be said, such a result of patients having something so visible in their blood would be a stunning rebuke to the doyens of “Institutional Medicine”–especially those in the UK and US governments, who have insisted that the cause of myalgic encephalomyelitis is so ~mysterious~ as to be practically unknowable! So unknowable they felt justified in neglecting to fund its research for these long decades…
References:
- Che X, Brydges CR, Yu Y, Price A, Joshi S, Roy A, Lee B, Barupal DK, Cheng A, Palmer DM, Levine S, Peterson DL, Vernon SD, Bateman L, Hornig M, Montoya JG, Komaroff AL, Fiehn O, Lipkin WI. Evidence for Peroxisomal Dysfunction and Dysregulation of the CDP-Choline Pathway in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. medRxiv [Preprint]. 2022 Jan 11:2021.06.14.21258895. doi: 10.1101/2021.06.14.21258895. PMID: 35043127; PMCID: PMC8764736.
- Che, X.; Brydges, C.R.; Yu, Y.; Price, A.; Joshi, S.; Roy, A.; Lee, B.; Barupal, D.K.; Cheng, A.; Palmer, D.M.; Levine, S.; Peterson, D.L.; Vernon, S.D.; Bateman, L.; Hornig, M.; Montoya, J.G.; Komaroff, A.L.; Fiehn, O.; Lipkin, W.I. Metabolomic Evidence for Peroxisomal Dysfunction in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2022, 23, 7906. https://doi.org/10.3390/ijms23147906
- Islinger M, Voelkl A, Fahimi HD, Schrader M. The peroxisome: an update on mysteries 2.0. Histochem Cell Biol. 2018 Nov;150(5):443-471. doi: 10.1007/s00418-018-1722-5. Epub 2018 Sep 15. PMID: 30219925; PMCID: PMC6182659.
- Bonekamp NA, Völkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors. 2009 Jul-Aug;35(4):346-55. doi: 10.1002/biof.48. PMID: 19459143.
- Antonenkov VD. Dehydrogenases of the pentose phosphate pathway in rat liver peroxisomes. Eur J Biochem. 1989 Jul 15;183(1):75-82. doi: 10.1111/j.1432-1033.1989.tb14898.x. PMID: 2753047.
- Wikimedia Foundation. Lorenzo’s Oil. Wikipedia. Retrieved July 12, 2022, from https://en.wikipedia.org/wiki/Lorenzo%27s_Oil
- Carillo MR, Bertapelle C, Scialò F, Siervo M, Spagnuolo G, Simeone M, Peluso G, Digilio FA. L-Carnitine in Drosophila: A Review. Antioxidants (Basel). 2020 Dec 21;9(12):1310. doi: 10.3390/antiox9121310. PMID: 33371457; PMCID: PMC7767417.
- Messias MCF, Mecatti GC, Priolli DG, de Oliveira Carvalho P. Plasmalogen lipids: functional mechanism and their involvement in gastrointestinal cancer. Lipids Health Dis. 2018 Mar 7;17(1):41. doi: 10.1186/s12944-018-0685-9. PMID: 29514688; PMCID: PMC5842581.
- Wanders RJ, Poll-The BT. “Role of peroxisomes in human lipid metabolism and its importance for neurological development”. Neurosci Lett. 2017 Jan 10;637:11-17. doi: 10.1016/j.neulet.2015.06.018. Epub 2015 Jun 18. PMID: 26095698.
- Wanders RJA, Waterham HR, Ferdinandusse S. Peroxisomes and Their Central Role in Metabolic Interaction Networks in Humans. Subcell Biochem. 2018;89:345-365. doi: 10.1007/978-981-13-2233-4_15. PMID: 30378031.
- Wikimedia Foundation. Glutamic acid. Retrieved July 12, 2022, from https://en.wikipedia.org/wiki/Glutamic_acid#Metabolism
- Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020 Jan 3;11(1):102. doi: 10.1038/s41467-019-13668-3. PMID: 31900386; PMCID: PMC6941980.
- Zhang Y, Zhang M, Zhu W, Yu J, Wang Q, Zhang J, Cui Y, Pan X, Gao X, Sun H. Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model. Redox Biol. 2020 Jan;28:101365. doi: 10.1016/j.redox.2019.101365. Epub 2019 Oct 31. PMID: 31707354; PMCID: PMC6854095.
- Ron Davis and Janet Dafoe. Ron Davis Updates on the Manganese Grant & Study! Retrieved July 14 from https://www.youtube.com/watch?v=Cf8_xz0bVZc
- Chander M, Setlow B, Setlow P. The enzymatic activity of phosphoglycerate mutase from gram-positive endospore-forming bacteria requires Mn2+ and is pH sensitive. Can J Microbiol. 1998 Aug;44(8):759-67. doi: 10.1139/cjm-44-8-759. PMID: 9830105.
- Wikimedia Foundation. SOD2. Retrieved July 12, 2022, from https://en.wikipedia.org/wiki/SOD2
- Missailidis D, Annesley SJ, Allan CY, Sanislav O, Lidbury BA, Lewis DP, Fisher PR. An Isolated Complex V Inefficiency and Dysregulated Mitochondrial Function in Immortalized Lymphocytes from ME/CFS Patients. Int J Mol Sci. 2020 Feb 6;21(3):1074. doi: 10.3390/ijms21031074. PMID: 32041178; PMCID: PMC7036826.
- Kennedy G, Spence V, Underwood C, Belch JJ. Increased neutrophil apoptosis in chronic fatigue syndrome. J Clin Pathol. 2004 Aug;57(8):891-3. doi: 10.1136/jcp.2003.015511. PMID: 15280416; PMCID: PMC1770396.
- Frémont, M., Vaeyens, F., Herst, C. V., De Meirleir, K., & Englebienne, P. (2006). Antiviral Pathway Deregulation of Chronic Fatigue Syndrome Induces Nitric Oxide Production in Immune Cells That Precludes a Resolution of the Inflammatory Response. Journal of Chronic Fatigue Syndrome, 13(4), 17–28. doi:10.1300/j092v13n04_03
- Hoel F, Hoel A, Pettersen IK, Rekeland IG, Risa K, Alme K, Sørland K, Fosså A, Lien K, Herder I, Thürmer HL, Gotaas ME, Schäfer C, Berge RK, Sommerfelt K, Marti HP, Dahl O, Mella O, Fluge Ø, Tronstad KJ. A map of metabolic phenotypes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight. 2021 Aug 23;6(16):e149217. doi: 10.1172/jci.insight.149217. PMID: 34423789; PMCID: PMC8409979.
- Naviaux RK, Naviaux JC, Li K, Bright AT, Alaynick WA, Wang L, Baxter A, Nathan N, Anderson W, Gordon E. Metabolic features of chronic fatigue syndrome. Proc Natl Acad Sci U S A. 2016 Sep 13;113(37):E5472-80. doi: 10.1073/pnas.1607571113. Epub 2016 Aug 29. Erratum in: Proc Natl Acad Sci U S A. 2017 May 2;114(18):E3749. PMID: 27573827; PMCID: PMC5027464.
- Hokama Y, Empey-Campora C, Hara C, Higa N, Siu N, Lau R, Kuribayashi T, Yabusaki K. Acute phase phospholipids related to the cardiolipin of mitochondria in the sera of patients with chronic fatigue syndrome (CFS), chronic Ciguatera fish poisoning (CCFP), and other diseases attributed to chemicals, Gulf War, and marine toxins. J Clin Lab Anal. 2008;22(2):99-105. doi: 10.1002/jcla.20217. PMID: 18348309; PMCID: PMC6649096.
- Hokama Y, Uto GA, Palafox NA, Enlander D, Jordan E, Cocchetto A. Chronic phase lipids in sera of chronic fatigue syndrome (CFS), chronic ciguatera fish poisoning (CCFP), hepatitis B, and cancer with antigenic epitope resembling ciguatoxin, as assessed with MAb-CTX. J Clin Lab Anal. 2003;17(4):132-9. doi: 10.1002/jcla.10079. PMID: 12784262; PMCID: PMC6807878.
- Amy Proal. ‘So it turns out we did find “something in the blood.”‘ https://twitter.com/microbeminded2/status/1534627222748815368 Retrieved July 12, 2022.
- Massimo Nunes, Arneaux Kruger, Amy Proal et al. The occurrence of hyperactivated platelets and fibrinaloid microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS), 22 June 2022, PREPRINT (Version 2) available at Research Square [https://doi.org/10.21203/rs.3.rs-1727226/v2]
- Pretorius E, Vlok M, Venter C, Bezuidenhout JA, Laubscher GJ, Steenkamp J, Kell DB. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. 2021 Aug 23;20(1):172. doi: 10.1186/s12933-021-01359-7. PMID: 34425843; PMCID: PMC8381139.
- Johnson, Hillary. Osler’s Web. Crown Publishing, 1996, p. 215.
- Simpson LO, Shand BI, Olds RJ. Blood rheology and myalgic encephalomyelitis: a pilot study. Pathology. 1986 Apr;18(2):190-2. doi: 10.3109/00313028609059457. PMID: 3093959.
- Simpson LO. Nondiscocytic erythrocytes in myalgic encephalomyelitis. N Z Med J. 1989 Mar 22;102(864):126-7. PMID: 2927808.
- Saha AK, Schmidt BR, Wilhelmy J, Nguyen V, Abugherir A, Do JK, Nemat-Gorgani M, Davis RW, Ramasubramanian AK. Red blood cell deformability is diminished in patients with Chronic Fatigue Syndrome. Clin Hemorheol Microcirc. 2019;71(1):113-116. doi: 10.3233/CH-180469. PMID: 30594919; PMCID: PMC6398549.
- Berg D, Berg LH, Couvaras J, Harrison H. Chronic fatigue syndrome and/or fibromyalgia as a variation of antiphospholipid antibody syndrome: an explanatory model and approach to laboratory diagnosis. Blood Coagul Fibrinolysis. 1999 Oct;10(7):435-8. doi: 10.1097/00001721-199910000-00006. PMID: 10695770.
- Kennedy G, Norris G, Spence V, McLaren M, Belch JJ. Is chronic fatigue syndrome associated with platelet activation? Blood Coagul Fibrinolysis. 2006 Mar;17(2):89-92. doi: 10.1097/01.mbc.0000214705.80997.73. PMID: 16479189.
2 thoughts on “Peroxisomes, Mitochondria, and Fibrinoid Microclots: Two New Preprints.”