
To donate in support of this research go here and select “Chronic Fatigue/ME Research” from the dropdown menu and indicate “for Professor Fischer’s ME research” in the comment box.
This is a short post based on a recent article I found that (at least where I looked) appears to have been overlooked by patient discussion groups.
Thanks to the field of metabolomics, we now know what many patients, researchers and physicians have long suspected: that metabolism in abnormal in myalgic encephalomyelitis (ME) [1, 2, 3, 4].
TLDR
In this article I review a very interesting and potentially very important study by Daniel Missailidis and colleagues on mitochondria function in myalgic encephalomyelitis versus healthy controls (link).
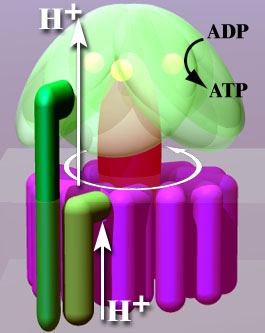
Though there were several interesting findings, the principal one was that ATP Synthase (aka Complex V; the enzyme the mitochondria use to transform electrochemical energy into usable energy) was nearly 25% inefficient in lymphoblasts derived from patients as compared to those from healthy controls. That is a lot. For instance, consider how tightly controlled pH is in the blood–with a range of only ~ .1. Most things in the body are likewise tightly controlled and I think if this result is representative of what is going on in our bodies our bodies will definitely “feel” that loss. Probably it would result in multiple systems being thrown off–considering how important cellular energy is to virtually all bodily functions.
What could be the cause? The article did not commit to any particular cause but there are a few possibilities: it could be a toxin (whether from the environment or produced inside the body) binding to ATP Synthase and blocking it, a non-toxic protein interaction blocking it, a modification to its structure by a regulatory protein, or decrease in the proton-motive force (the electrochemical gradient which powers the mitochondria’s production of energy).
Another possibility is that this finding is due to the authors’ method of generating cells to test. They took lymphocytes (a type of immune cell) from the blood of healthy controls and ME patients and then used Epstein-Barr Virus (EBV) to transform these cells into lymphoblasts-the cells they tested. It is possible (perhaps due to ongoing infection, or to ongoing inflammation or even genetic susceptibility) that lymphocytes of ME patients react differently to the infection with EBV compared to control cells and this different reaction accounts for the differences in mitochondrial function observed in the tested cells. If this was the case it would (imo) not be as exciting as the earlier possibilities but nevertheless could still shed light on the nature of the immune dysfunction in myalgic encephalomyelitis.
Some Recapitulation
“Additional studies are required to identify the substance(s) in ME/CFS serum that mediate the effects on cultured muscle cells, which could act directly on the metabolic apparatus or indirectly via signaling factors.”
– Fluge et al., 2016.
One of the earlier, and most well-known, metabolic findings in myalgic encephalomyelitis is Robert Naviaux’s group’s finding that the metabolome (whole body of metabolites) of ME patients resembled a state of dauer (an alternative development state of C. elegans worms go into during environmental stress). While many pathways were abnormal, most pathways showed a decrease in metabolites “consistent with a hypometabolic syndrome” [1].
Another study by Neil McGregor, Chris Armstrong, and colleagues found that fall of the purine metabolite hypoxanthine concentration was associated with (self-reported) post-exertional malaise [2]. Secondly, they found this decrease correlated with an altered ratio of glucose to lactate (produced when the body is using anaerobic metabolism): suggestive of a glycolytic anomaly [2]. Thirdly, they found exertion of urine metabolites which indicated a hypermetabolic event (presumably due whatever exertion caused the post-exertional malaise) [2].
A recent study by Frederick and August Hoel, Fluge and Mella, and colleagues found two major and one minor metabolic subsets in ME patients: one major subset was associated with a lipolytic state where the body appeared to be burning fats to excess to compensate for a deficiency in carbohydrate (sugar) metabolism and a second major subset where there was increased fatty acid mobilization but potentially compromised fatty acid oxidation [3].
An earlier study by Fluge, Mella and colleagues postulated an impairment in pyruvate dehydrogenase (an important enzyme for transferring pyruvate, a product of glycolysis, to the mitochondria) based on a reduction of amino acids which fuel oxidation in the Tricarboxylic Acid Cycle (in the mitochondria) mainly in female patients [4]. In male patients they found an increased marker of protein catabolism [4]. Also, similarly to the results of Ron Davis’s group, [5] myoblasts (muscle precursor cells) Fluge and Mella grew in myalgic encephalomyelitis patient serum displayed abnormal metabolic features: including increased oxidative respiration and lactate secretion [4] suggesting a factor in patients’ blood which supports respiration.
As these few studies demonstrate there is a wealth of evidence that something is wrong in metabolism in ME and, that the mitochondria are, probably, involved somehow. However, when it comes to what exactly is wrong the studies do not completely agree. This may be due to many factors: studies may be using patient groups selected by different definitions (though these five studies all used the Canadian Criteria [6]), also they may have used patients with different ages, different gender distribution, different lengths of illness and different initiating factors. And of course, another factor is the studies all used differing methods for measuring the concentration of metabolites. Lastly, most myalgic encephalomyelitis studies tend to use smaller sample populations: something we should blame our respective governments and grant-funding agencies for…
The Mitochondrion: A Logical Starting Point
If one were to look for a place in the body which could be a cause of abnormal energy metabolism, everyone’s first guess would be the mitochondrion. This is because the mitochondria are responsible for the most important process of energy acquisition: the oxidation of carbon-chains to produce carbon-dioxide and water (not entirely dissimilar from how a furnace burns wood to produce smoke and heat).

In this study, “An Isolated Complex V Inefficiency and Dysregulated Mitochondrial Function in Immortalized Lymphocytes from ME/CFS Patients,” Daniel Missailidis and colleagues aimed to measure the metabolic effectiveness of lymphocytes from ME patients as compared those from healthy controls [7]. Firstly, the cells were converted to lymphoblasts and then had their energetic and mitochondrial function measured with various assays, including Seahorse extracellular flux analysis [7].
Many abnormalities were found!
“The inefficiency of ATP synthesis by Complex V means that basal respiration rates by ME/CFS lymphoblast mitochondria would also be reduced, were it not for the compensatory upregulation of their respiratory complex levels. This allows them to maintain normal ATP synthesis rates and, as observed, is accompanied by increased respiratory capacity of the electron transport chain (mostly Complex I activity), supported by an increased use of the proton gradient to drive mitochondrial protein import and other mitochondrial membrane transport processes (the “proton leak”). ”
– Daniel Missailidis et al., 2020.
Perhaps most significantly, the authors found that the as a proportion of baseline oxygen consumption rate the rate of ATP synthesis was 15% decreased compared to controls [7]. This would seem to directly implicate the ATP Synthase (aka. Complex V) in whatever disease process is occuring in ME.
Next, the authors looked for increases in the other enzymes which comprise the electron transport chain needed to power ATP Synthase (this is by the creation of a proton gradient between the inner mitochondrial membrane and the outer mitochondrial membrane) and found increases in subunits of complexes I, II, and IV (Including, the ATP Synthase there are five complexes). They also found an increased maximum oxygen-consumption rate when the ME patient’s mitochondria were chemically uncoupled [7]. These findings indicate that the mitochondria of ME patients are working overtime to produce energy in the presence of some acquired defect: much like if you broke your leg and walked on crutches your arms and good leg would get stronger.
A proteomics analysis of the mitochondrial respiratory chain complexes confirmed the earlier findings and, due to an enhanced concentration of Complex V in the ME patient lymphoblasts, caused the authors to reassess the ATP Synthase inefficiency to almost 25% [7].
Surprisingly to many of us, the level of intracellular reactive oxygen species was not higher in myalgic encephalomyelitis-derived lymphoblasts [7]. However, mitochondrial membrane potential was reduced in ME/CFS cells, potentially a result of the inefficiency of ATP synthesis causing an increased use of the proton gradient to drive membrane transport processes according to the authors [7].
Fatty Acid Synthesis Abnormalities
Abnormal elevations were also found in fatty acid transport proteins ( carnitine acyltransferase I, II, acyl carrier protein) and 17 of 20 proteins involved in Beta-Oxidation (fatty acid oxidation) [7]. Taken together this would seem to indicate that the cells of ME patients have increased their fatty acid catabolism and electron transport chain proteins in an effort to wring more energy out of a faulty motor-analogous to pressing hard down on the pedal when your car’s engine is sputtering.
mTORC1
mTORC1, Mammalian Target of Rapamycin Complex I, is a protein complex involved in the control of translation (the creation of proteins from RNA templates) of proteins. The authors found that E4-BP1 a protein involved in translation, was significantly more phosphorylated (in this case meaning activated) in ME patient-derived lymphoblasts indicating increased activity of mTORC1 [7].
More ATP Synthase/Complex V
In a disease in which exertion makes the patients worse and one of the main patient reports is a lack of energy both mental and physical mitochondria are perhaps the most obvious: target followed by the heart and circulatory system. If Missailidis and colleagues’ study is correct, not only is there a defect at this target but the abnormality affects one of the most integral mitochondrial components–the ATP synthase.
The most immediate question suggested by this study is: what is causing the ATP synthase to be inefficient? One option, that the proton-motive force is less in ME patient mitochondria is mentioned by the authors as a potential cause [7]. While study did find the inner mitochondrial membrane potential was less in ME-derived lymphoblasts the authors point out the unused respiratory capacity of ME-derived lymphoblasts should be sufficient to allow ATP Synthase to operate at normal capacity [7]. Other potential causes exist: there could be toxin binding (for instance, polyenic α-pyrone derivatives from some molds [8]) to the ATP-synthase, there could be a protein interaction [7] causing it to be inhibited (such as ATPIF1 or Angiostatin [8, 9]) or or there could be an inhibitory modification to its structure. It is even possible that the aberrant concentration of some metabolite results in a negative feedback resulting in ATP-synthase inhibition. Considering the importance of ATP Synthase to life, the question of what is repressing its efficiency in ME will, hopefully, draw great research interest.
It is also noteworthy that the authors used EBV (Epstein-Barr Virus) to transform patient-derived and healthy control-derived lymphocytes into lymphoblasts [7]. It is possible, especially if myalgic encephalomyelitis is caused by a chronic infection, viral reactivation, or immune response of some kind, that patients’ cells would react to this transformation in a different way compared to HC cells. Unfortunately, I am not knowledgeable enough to be able to state if the former is likely or very unlikely: if it is likely hopefully further studies can be done to rule out this possibility.
More Relevant Studies
One study which might be relevant is a study by Philipp Schreiner, Robert Naviaux, Bhupesh Prusty and colleagues which showed that when cells with an integrated HHV-6 (a herpesvirus heavily implicated in the initial phase of ME) were infected with a new virus they secreted a factor that fragmented mitochondria [10]. This factor acted similarly to serum from ME patients (which also had this effect).
Along the same lines, in his nanoneedle study, Ron Davis and colleagues found that when cells from ME patients were incubated in blood plasma from patients they displayed an abnormal impedance (a quality of alternating electrical circuits similar to resistance ) but with blood plasma from controls the cells instead had a normal impedance [5]. This is strong evidence there is some factor(s) in the plasma which is driving the “ME cell phenotype” [4, 5]. As best as I could determine, the identity of this factor is unfortunately as yet unknown.
There is also more direct evidence that something is going wrong with the mitochondria-and ATP synthase specifically: A study by Eiren Sweetman, Warren Tate, Rosamund Vallings and colleagues found, using a mass-spectroscopy analysis that many proteins involved in mitochondrial function were upregulated [11]. Specifically, they found proteins in the immediate pathways upstream of ATP Synthase were upregulated and conclude: “results from this study support a model of deficient ATP production in ME/CFS, compensated for by upregulation of immediate pathways upstream of Complex V that would suggest an elevation of oxidative stress” [11].
It is fair to say that the results of several groups have converged on the mitochondria as site affected by the disease, and perhaps even directly downstream of the causal mechanism. These studies are surprisingly congruent, more so than is usually found in other areas of research. I hope very much that studies in the coming months and years can identify the factor which causes ME cells to adopted a “diseased” phenotype and the factor (possibly the same, almost certainly related) which causes the abnormality in ATP synthase in the mitochondria.
My Concluding Thoughts
It used to be said that nothing much is wrong in ME patients because the standard lab tests usually returns normal except for low Erythrocyte SED rate. However the problem in recent “omic” research is that too much is wrong and the studies (for instance mRNA studies or metabolomics studies) [1-4] often don’t seem to agree with each other on which is the major thing and which things are mere downstream effects. If there is a problem in the mitochondria (and ATP-synthase especially), given their centrality to the cell’s energy production, (and thus to virtually all the functions of life) this would also have the effect of simplifying things–since it is likely whatever is impairing the mitochondria is either a direct cause or closely proximate to the pathomechanism of myalgic encephalomyelitis…
Quotes of Interest from the Study
Our results showed that ME/CFS lymphoblasts exhibit an isolated Complex V inefficiency that is accompanied by upregulation of mitochondrial protein expression, including mitochondrial respiratory complexes and enzymes involved in the TCA cycle, fatty acid uptake and β-oxidation. These findings confirm that these ME/CFS cells do indeed exhibit a mitochondrial deficiency in ATP generation, but reveal that, in lymphoblasts at least, this specifically involves Complex V rather than a generalized reduction in all mitochondrial functions.
“We found that not only were PBMCs quiescent, but the fraction of dead cells after 1–3 days incubation in culture medium was dramatically greater for ME/CFS lymphocytes than for control lymphocytes. It is likely that in some previous studies, the ME/CFS lymphocytes assayed for mitochondrial activity included a higher proportion of dead cells than did the controls.”
It is possible that the dysregulation of Complex V and mitochondrial function in ME/CFS lymphoblasts arises because they respond differently to EBV infection than do control lymphoblasts (…) it is possible that in ME/CFS lymphoblasts, EBV reactivates more readily to enter the lytic cycle and this in turn affects mitochondrial function.
Previous steady state measurements and metabolic flux measurements of mitochondrial respiratory function in ME/CFS lymphocytes have suggested that in ME/CFS cells there is either a generalized reduction [6,7,8] or little change [9,15] in mitochondrial activity and respiratory capacity. However, functionally normal OXPHOS Complex I to IV activity has also been reported in ME/CFS lymphocytes [11,41], while the expression of mitochondrial proteins is upregulated in patient saliva, platelets and lymphocytes [12,13,23]. Elevated nonmitochondrial ATP production has also been reported in ME/CFS lymphocytes [11].
In this work we have resolved these inconsistencies by revisiting the issue of mitochondrial function and capacity in immortalized lymphocytes (lymphoblastoid cell lines or lymphoblasts).
It is known that mitochondrial ATP synthase activity can be regulated by a variety of proteins, small molecules and signaling pathways, some of them by acting through Complex V’s own inhibitory subunit AIF1 [53,54]. These possible causes for Complex V inefficiency in ME/CFS lymphoblast mitochondria should be investigated in future work.
However, despite lower steady state ATP levels, the AMPK activation state was not significantly different between cultured muscle cells from CFS patients (Fukuda criteria) and healthy controls [62]. Future work should therefore test the hypothesis that AMPK is chronically activated in ME/CFS lymphoblasts and other cell types.
Together our results suggest a model in which the Complex V defect is a proximal activator of compensatory upregulation of expression of mitochondrial proteins.
Most notable amongst the mitochondrial proteins whose translation is upregulated by TORC1 are nuclear-encoded subunits of Complexes I and V [20], the two respiratory complexes whose expression we found to be the most evidently elevated in the whole cell proteomes of ME/CFS lymphoblasts.
It would be valuable in future experiments to measure fatty acid utilization rates in ME/CFS and control lymphoblasts.
References:
- Naviaux RK, Naviaux JC, Li K, Bright AT, Alaynick WA, Wang L, Baxter A, Nathan N, Anderson W, Gordon E. Metabolic features of chronic fatigue syndrome. Proc Natl Acad Sci U S A. 2016 Sep 13;113(37):E5472-80. doi: 10.1073/pnas.1607571113. Epub 2016 Aug 29. Erratum in: Proc Natl Acad Sci USA. 2017 May 2;114(18):E3749. PMID: 27573827; PMCID: PMC5027464.
- McGregor NR, Armstrong CW, Lewis DP, Gooley PR. Post-Exertional Malaise Is Associated with Hypermetabolism, Hypoacetylation and Purine Metabolism Deregulation in ME/CFS Cases. Diagnostics (Basel). 2019 Jul 4;9(3):70. doi: 10.3390/diagnostics9030070. PMID: 31277442; PMCID: PMC6787670.
- Hoel F, Hoel A, Pettersen IK, Rekeland IG, Risa K, Alme K, Sørland K, Fosså A, Lien K, Herder I, Thürmer HL, Gotaas ME, Schäfer C, Berge RK, Sommerfelt K, Marti HP, Dahl O, Mella O, Fluge Ø, Tronstad KJ. A map of metabolic phenotypes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight. 2021 Aug 23;6(16):e149217. doi: 10.1172/jci.insight.149217. PMID: 34423789; PMCID: PMC8409979.
- Fluge Ø, Mella O, Bruland O, Risa K, Dyrstad SE, Alme K, Rekeland IG, Sapkota D, Røsland GV, Fosså A, Ktoridou-Valen I, Lunde S, Sørland K, Lien K, Herder I, Thürmer H, Gotaas ME, Baranowska KA, Bohnen LM, Schäfer C, McCann A, Sommerfelt K, Helgeland L, Ueland PM, Dahl O, Tronstad KJ. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. 2016 Dec 22;1(21):e89376. doi: 10.1172/jci.insight.89376. PMID: 28018972; PMCID: PMC5161229.
- Esfandyarpour R, Kashi A, Nemat-Gorgani M, Wilhelmy J, Davis RW. A nanoelectronics-blood-based diagnostic biomarker for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Proc Natl Acad Sci U S A. 2019 May 21;116(21):10250-10257. doi: 10.1073/pnas.1901274116. Epub 2019 Apr 29. PMID: 31036648; PMCID: PMC6535016.
- Carruthers B.M., Jain A.K., De Meirleir K.L., Peterson D.L., Klimas N.G., Lerner A.M., Bested A.C., Flor-Henry P., Joshi P., Powles A.C., et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Clinical Working Case Definition, Diagnostic and Treatment Protocols. J. Chronic Fatigue Syndr. 2003;11:7–36. doi: 10.1300/J092v11n01_02.
- Missailidis D, Annesley SJ, Allan CY, Sanislav O, Lidbury BA, Lewis DP, Fisher PR. An Isolated Complex V Inefficiency and Dysregulated Mitochondrial Function in Immortalized Lymphocytes from ME/CFS Patients. Int J Mol Sci. 2020 Feb 6;21(3):1074. doi: 10.3390/ijms21031074. PMID: 32041178; PMCID: PMC7036826.
- Hong S, Pedersen PL. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiol Mol Biol Rev. 2008 Dec;72(4):590-641, Table of Contents. doi: 10.1128/MMBR.00016-08. PMID: 19052322; PMCID: PMC2593570.
- García-Bermúdez J, Cuezva JM. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim Biophys Acta. 2016 Aug;1857(8):1167-1182. doi: 10.1016/j.bbabio.2016.02.004. Epub 2016 Feb 12. PMID: 26876430.
- Schreiner P, Harrer T, Scheibenbogen C, Lamer S, Schlosser A, Naviaux RK, Prusty BK. Human Herpesvirus-6 Reactivation, Mitochondrial Fragmentation, and the Coordination of Antiviral and Metabolic Phenotypes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Immunohorizons. 2020 Apr 23;4(4):201-215. doi: 10.4049/immunohorizons.2000006. PMID: 32327453.
- Sweetman E, Kleffmann T, Edgar C, de Lange M, Vallings R, Tate W. A SWATH-MS analysis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome peripheral blood mononuclear cell proteomes reveals mitochondrial dysfunction. J Transl Med. 2020 Sep 24;18(1):365. doi: 10.1186/s12967-020-02533-3. PMID: 32972442; PMCID: PMC7512220.
Thank you so much for the great summary here.
I keep thinking that to find out if the mitochondrial dysfunctions are up or downstream etc, one way to build up a picture would be compare ME with the metabolomics from other patient groups; of which there should be some in literature already:
Spinal cord injury patients.
Thromboembolism
Viral complications eg viral encephalitis, HIV, ??
Autoimmune involving energy metabolism
Classical mitochondrial diseases
I come at this from a different angle since my fatigue and symptoms are almost lifelong but gradually worsened especially at puberty. I think mitochondrial insufficiency might have always been there in my case, amplified by whatever stressors and viral situations came along.
Not the case obviously for ME in a previously athletic adult 😂
Thanks!
I also think it would be worthwhile to compare the whole body of metabolites of people with ME to various other diseases and look for a close match. The difficulty as you said is that for the comparison to be meaningful you would need, at minimum, several other diseases/states of altered metabolism to compare to. For instance, just focusing on diseases of metabolism, you would probably want to compare the metabolome in ME patients to the metabolome in patients with disorders of glycolysis, disorders in pyruvate transport to the mitochondria, disorders in the Krebs Cycle, disorders of ATP-Synthase. And that is not including any disease controls for chronic inflammation from autoimmunity such as MS, or from an infection such as HIV.
Another difficulty is it seems like the body of metabolites in ME changes post-exertion and so it would be best if studies could quantify if they were measuring the patient’s metabolites in a state of rest, a state of mild exertion, moderate exertion etc. Probably for most of the studies the patients had to at least go to the labs to get their blood drawn–which, for me, would be significant exertion!
I think two ways around the first problem would be:
1) if you knew very precisely what you were searching for (for instance, in the Missailidis study: ATP Synthase) or
2) if there was a standardized database of the metabolome in different diseases (I don’t know if this exists yet). I think part of the issue is that different labs are using at least slightly different ways to measure metabolites: for instance even the metabolite studies in ME do not completely agree with each other. Ron Davis summarized these studies like this “People [patients] are not burning glucose very well, and the other thing is people are not burning fats very well. But they do burn amino acids and that’s been pretty consistent”
Re: Onset I do know at least one patient who also got sick as a very young child, though it doesn’t seem as common as people getting sick in their teenage or young adult years.