
Dumping Recyclables

After wrestling with this problem for many years, the only explanation I can come up with is that the cumulative sum of blood vessel diameter is reduced. The pipes are smaller than they should be.
David Bell, MD. Former Chairman of the CFSCC and chief investigator of the Lyndonville ME Outbreak.
Do you ever have the feeling of being spaced out? Thoughts swirling around in your head but none of them is willing to land and materialize? (At least I hope there are thoughts in my head–we shall see) So in lieu of a longer post here is a short theory I’ve been thinking about…
One thing I’ve always found odd is that at times the disease seems to fade, not entirely but by a sizable amount. Usually, those times are when I would expect myself to be having a rush of adrenaline. Adrenaline (also known as epinephrine) is the classic fight-or-flight hormone and perhaps due to that reason it is much easier to link a conscious state of being with a spike in it as compared to other hormones like growth hormone or follicle stimulating hormone. It has two major effects: firstly it dilates blood vessels going to skeletal muscle and vital organs such as the heart, and secondly it suppresses inflammation. The second effect explains why the most common form of adrenaline we will encounter in everyday life is the EpiPen for example when someone accidentally eats peanut residue in their chicken.
There are reams of evidence that inflammation is increased in myalgic encephalomyelitis. For instance, the ME research group found elevated isoprantanes (a marker of oxidative stress)1 in patients and earlier found increased neutrophil apoptosis (cell death)2. There are increased cytokine mRNA3 after exercise and another study found symptoms tend to correlate with the levels of the hormone leptin4. Altered red blood cell deformability5 (likely the result of oxidative stress5) and altered morphology6 has also been found in ME patients. Thus for starting, it seems reasonable to suppose inflammation is some way driving the horrible physical and cognitive symptoms. The short periods of feeling better when in fight or flight mode and the immune system is suppressed provide circumstantial evidence seemingly in support of this idea. Though I have never seen a formal study on it, pregnancy is also famously a common anecdotal period of symptom waning in some persons. If the immune system is at fault in terms of generating inflammation then that leads to two immediate questions:
1.) What is the immune system doing to cause the symptoms?
2.) Why is the immune system acting in this way?
For the first question, I do not think I can make a definite guess beyond that it is blocking the body’s energy production in some direct way. Considering the available research, it would make sense this is either through an inhibitory effect on the mitochondria or through a circulatory mechanism such as blood vessel integrity or damage to red blood cells. This guess assumes the energy deficit we experience is both systemic in nature and “physiologically real”–not, for instance, an effect of the nervous system causing the subjective experience of exhaustion.
As for the second question, it makes sense to postulate there is some noxious mediator in the body causing the immune system to overreact. Usually this is discussed in terms of autoimmunity versus chronic infection but I believe another possibility is a metabolic toxin or cytotoxin which the body simply doesn’t effectively remove–hence the title. A transient block of the immune system most elegantly explains the short periods of hours or days where people have dramatically reduced symptoms which like clockwork reappear with any signs of ever having been interrupted.
In the two flagship theories of myalgic encephalomyelitis, autoimmunity and chronic infection a transient block of the immune system would also have the same effect of a period of reduced symptoms but one would expect at least occasionally these periods of reduced symptoms would be followed by dramatic progressions of the disease (from the organism spreading to more tissues, or from the immune system reacting violently to new antigens) and this doesn’t seem to happen. However, since very little is known about the pathology of ME it may be that the infection is an abortive one, or that the autoimmune reaction is to some marker which has high rate of turnover (and therefore never buildups past a certain point). Therefore the autoimmunity, infection and noxious stimulant models can potentially account for this finding of transient reduction of symptoms following a fight-or-flight situation.
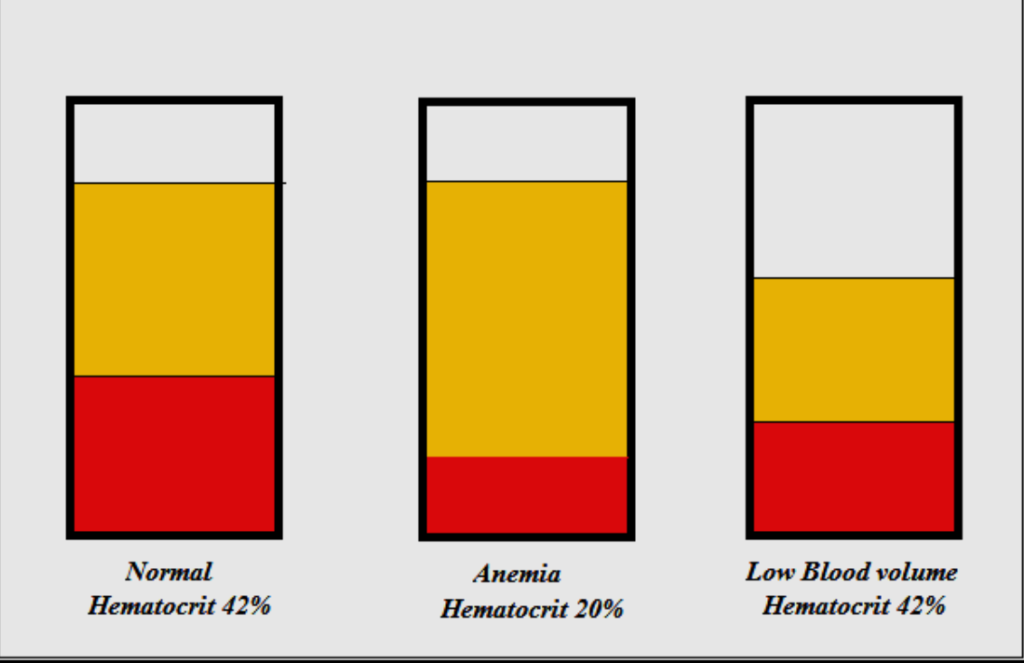
2nd: Person with Anemia having reduced fraction of blood cells.
3rd: A Person with ME with reduced blood plasma and reduced blood cells.
To return to Dr. Bell’s quote one of the most ironclad findings in myalgic encephalomyelitis is the abnormal hemodynamic response of patients to upright posture and the loss of blood volume, principally RBC volume, seen in at about 2/3’s of patients 7,8,9 . The ME research UK team also found that there is an abnormal prolonged vasodilation10 in the microcirculation of patients. Taken together these and other results would seem to pinpoint the circulatory system as important, and perhaps principal locus of abnormalities in ME. Are these abnormalities a primary driver of the disease or only a secondary effect?
While we don’t know it is tempting to imagine a protein or fat (a noxious mediator) that is present in an abnormal quantity or configuration that causes loss of cellular respiratory capability either through the immune response, or directly and that, again either through the immune response or directly causes heightened inflammation but not its effective removal. This basic setup could explain several bizarre occurrences in ME. For instance, if the immune response is a driver of symptoms and disease then the immune suppression in pregnancy could be expected to often result in a reduction of symptoms. For the same reason, there would be a reduction of symptoms in cases of fight-or-flight where there is a high release of adrenaline.
This hypothetical noxious mediator could form a disease-sustaining feedback loop if it induces either inflammation or induces effects, such a loss of cellular respiratory capacity which lead to the body being unable to get rid of the original mediator. Most of the signalling molecules in our body are short-lived, minutes, hours, days–they rarely stick around. However if there was a mediator that induced dramatic effects such as cell damage or inflammation it is possible that in susceptible persons that mediator might shut-down the cleanup system designed to remove it to the extent it persists at a much higher concentration than in health. This then is the crux of the Dumped Recyclable Model. For one concrete illustration, one could imagine there is oxidative damage to the red blood cells that lead to them inducing inflammation when traveling through vascular tissues but, for whatever reason, not their prompt removal by the spleen.
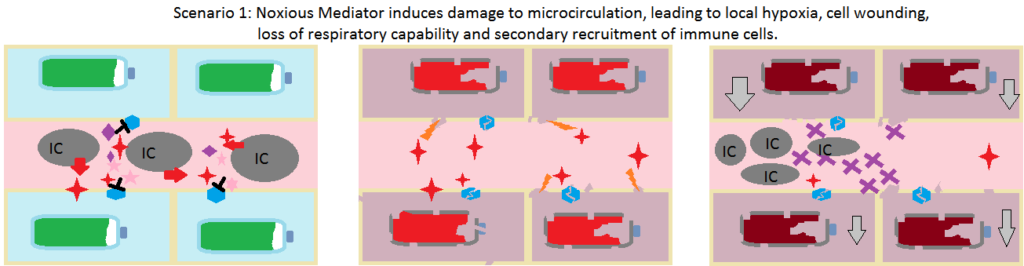
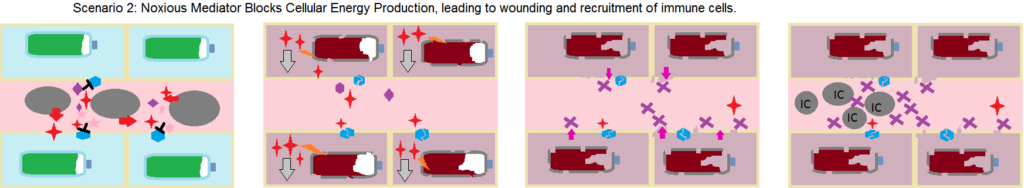

Another question is: what would this model predict directly causes the impaired metabolic state in ME? It could be that inflammation directly causes the loss of respiratory capacity most probably due to local blood flow disruption, less likely due to inflammatory mediators signalling the cell to slow respiration, or secondly it could be that the noxious stimulant itself induces an arrest of cellular respiration or harms cellular respiratory capacity in some way, these effects then leading to inflammation.
To sum up, the dumped recyclable model would contain three core components:
1.) A endogenous molecule(s) which under normal circumstances is recycled but in susceptible people the recycling pathway fails causes the molecule to be “dumped” and build up.
2.) A state of constitutively high inflammation or hypersensitive induction of inflammation.
3.) A state of diminished metabolic capacity.
A simple picture of the disease then might be a person gets a transient state of high inflammation from an acute infection such as the flu. In this state of high inflammation a noxious stimulant is produced which in susceptible persons is not readily removed. This stimulant indirectly or directly stimulates the activation of the immune system leading to loss of metabolic capacity at the cellular level. (There is indirect evidence which implicates altered control of blood flow and/or altered permeability of small blood vessels.). This loss of metabolic capacity or a parallel side effect of the activated immune system leads to the recycling process for the noxious stimulant molecule to fail, leading it to build up and lead to an altered constitutional state of inflammation and metabolism.
Of course this process could still occur and still be important if the core feature of myalgic encephalomyelitis turns out to be a chronic infection or autoimmunity but in these cases this process would be only a secondary effect instead of the primary driver it would be in the noxious stimulant/mediator theory.
Perhaps I have only been recycling the same clouds through my mind. But if not, I hope you found these ideas worthwhile. In future health.
–Johnny Duncan
Postscript:
In what appears to me to be a seminal study, Frederick and August Hoel (in collaboration with Fluge, Mella, and others) identified two main metabolic phenotypes in Myalgic Encephalomyelitis by metabolomic profiling11. Both subtypes had features suggesting a disorder of cellular metabolism. One phenotype had features which suggested a lipolytic metabolic state (for instance, increased ketone bodies) and the other had features suggesting disrupted control of lipid metabolism11. In both cases, it appears oxidative respiration is a key chokepoint for whatever in this disease.
Citations:
- Kennedy G, Spence VA, McLaren M, Hill A, Underwood C, Belch JJ. Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med. 2005 Sep 1;39(5):584-9. doi: 10.1016/j.freeradbiomed.2005.04.020. PMID: 16085177.
- Kennedy G, Spence V, Underwood C, Belch JJ. Increased neutrophil apoptosis in chronic fatigue syndrome. J Clin Pathol. 2004 Aug;57(8):891-3. doi: 10.1136/jcp.2003.015511. PMID: 15280416; PMCID: PMC1770396.
- Light AR, Bateman L, Jo D, Hughen RW, Vanhaitsma TA, White AT, Light KC. Gene expression alterations at baseline and following moderate exercise in patients with Chronic Fatigue Syndrome and Fibromyalgia Syndrome. J Intern Med. 2012 Jan;271(1):64-81. doi: 10.1111/j.1365-2796.2011.02405.x. Epub 2011 Jul 13. PMID: 21615807; PMCID: PMC3175315.
- Stringer EA, Baker KS, Carroll IR, Montoya JG, Chu L, Maecker HT, Younger JW. Daily cytokine fluctuations, driven by leptin, are associated with fatigue severity in chronic fatigue syndrome: evidence of inflammatory pathology. J Transl Med. 2013 Apr 9;11:93. doi: 10.1186/1479-5876-11-93. PMID: 23570606; PMCID: PMC3637529.
- Saha AK, Schmidt BR, Wilhelmy J, Nguyen V, Abugherir A, Do JK, Nemat-Gorgani M, Davis RW, Ramasubramanian AK. Red blood cell deformability is diminished in patients with Chronic Fatigue Syndrome. Clin Hemorheol Microcirc. 2019;71(1):113-116. doi: 10.3233/CH-180469. PMID: 30594919; PMCID: PMC6398549.
- Simpson LO. Nondiscocytic erythrocytes in myalgic encephalomyelitis. N Z Med J. 1989 Mar 22;102(864):126-7. PMID: 2927808.
- Streeten DHP, Bell DS. Circulating blood volume in chronic fatigue syndrome. J Chronic Fatigue Syndr. 1998;4(1):3-11. https://doi.org/10.1300/J092v04n01_02
- Bell DS. Blood Volume in ME. Invest in ME Research. Presented 2015. http://www.investinme.org/Documents/Research/David%20Bell%20Blood%20Volume%20in%20ME%20Sep%2015.pdf
- Streeten DH, Thomas D, Bell DS. The roles of orthostatic hypotension, orthostatic tachycardia, and subnormal erythrocyte volume in the pathogenesis of the chronic fatigue syndrome. Am J Med Sci. 2000 Jul;320(1):1-8. doi: 10.1097/00000441-200007000-00001. PMID: 10910366.
- Spence VA, Khan F, Kennedy G, Abbot NC, Belch JJ. Acetylcholine mediated vasodilatation in the microcirculation of patients with chronic fatigue syndrome. Prostaglandins Leukot Essent Fatty Acids. 2004 Apr;70(4):403-7. doi: 10.1016/j.plefa.2003.12.016. PMID: 15041034.
- Hoel F, Hoel A, Pettersen IK, Rekeland IG, Risa K, Alme K, Sørland K, Fosså A, Lien K, Herder I, Thürmer HL, Gotaas ME, Schäfer C, Berge RK, Sommerfelt K, Marti HP, Dahl O, Mella O, Fluge Ø, Tronstad KJ. A map of metabolic phenotypes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight. 2021 Aug 23;6(16):e149217. doi: 10.1172/jci.insight.149217. PMID: 34423789; PMCID: PMC8409979.
Be the first to reply