
(Part 2 will be shorter)
A map of metabolic phenotypes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. 1 Lead Authors: Fredrik and August Hoel.
I believe this new paper from Norway (including the well-known Fluge and Mella as coauthors) is a seminal contribution to the understanding of the metabolic derangement which is now almost-universally acknowledged to exist in myalgic encephalomyelitis but the precise characteristics of which are, rightly, still debated.
On 83 myalgic encephalomyelitis patients vs. 35 controls the authors performed a global metabolomic panel as well as a lipidomic panel and hormone measurements. Though I might be incorrect on this, I think the lipidomic component is novel to this study.
As I mentioned in my petition, analysis of the lipidome is (the set of the total lipids in a the body or in a type of cell) is a sorely needed area of ME research. Lipids (aka fats) are obviously a very essential component to the process of making cellular energy since they can be burned by the mitochondria for higher energy returns compared to things like sugars. Therefore any disease which has or is suspected to have an impaired metabolism would to well to have patients’ lipidome investigated. In our disease, as well, there were the tantalizing findings of Professor Yoshitsugi Hokama, who found a lipid epitope which antigenically mimicked Ciguatera Toxin 2 (a lipophilic toxin which acts binding and forcing open the voltage-gated Na+ channel on nerve cells) overrepresented in our disease. These epitopes were later found to be phospholipids associated with cardiolipin from the mitochondria 3 –something which should be ordinarily shielded from the immune system. Lastly, many inflammatory mediators, such as arachidonic acid and prostaglandins, are derived from lipids. There is a large body of research indicating heightened inflammation in ME, most especially after exertion.4-8
Using statistical methods (k-means clustering followed by principal component analysis) on the panel of 880 metabolites measured Noel and Noel found the myalgic encephalomyelitis patients clustered into two major metabolic phenotypes and one minor phenotype.
There seemed to be several important findings. Both of the two metabolic phenotypes were associated with altered energy metabolism. Additionally there seemed to be some molecules ( e.g glycerol ) which were abnormal in both subtypes, which the authors suggest could be related to a core mechanism of ME disease. There were other metabolite molecules which were only abnormal in one subtype or another.
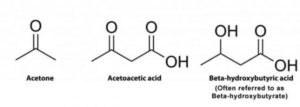
Subtype ME-M1 had elevated ketone bodies, high numbers of circulating fatty acid derivatives and decreased levels lipid compounds including: phospholipids and lysophospholipids, bile products, and sterols. ME-M1 also had low levels of several amino acids such as tryptophan, leucine/isoleucine/valine, arginine/proline, and phenylalanine and elevated lysine. Overall the authors propose ME-M1 represents a lipolytic state, with the body depending to an enhanced degree on fatty-acid catabolism to compensate for a (hypothesized) deficiency in burning carbohydrates for cellular respiration.
In contrast to subtype one subtype ME-M2 had lower level of fatty acid derivatives but increased fatty acid mobilization based on having an elevated glycerol (as with subtype M1). There was an increase in serum sucrose (a carbohydrate). Serum pyruvate was also elevated–possibly indicative of a defect in pyruvate oxidation. Metabolites of amino acids (the building blocks of proteins) tended to be increased and decreased in ME-M2, depending on the specific amino-acid. In contrast to subtype ME-M1, tryptophan was increased in serum but several of its downstream intermediates were decreased. Also in contrast to ME-M1 bile products were elevated in ME-M2. The authors propose ME-M2 could represent a state of disrupted control of lipid metabolism (in contrast to ME-M1 representing a deficiency in carbohydrate oxidation) with comprised lipid oxidation by the mitochondria.
Overall this study offers new impetus into looking at both the core mechanism causing disease in myalgic encephalomyelitis and the fact there may be downstream subtypes which are different enough to be clinically significant. A common theme was a posited derangement in the oxidation of materials by the mitochondria to produce ATP. In one subtype (ME-M1) the authors propose there is a dysfunction in the burning of carbohydrates (sugars) by the mitochondria, necessitating a shift to burning of fats. In the second subtype (ME-M2), the authors propose there is a dysfunction in the burning of fats by the mitochondria.
I think there are three ideas advanced about the nature of the disease myalgic encephalomyelitis by this paper that especially merit expansion and further study.
- That there is a common core pathogenic process in people with myalgic encephalomyelitis which, at minimum, exerts detectable metabolic effects.
- That there are subtypes which represent differing metabolic derangements superimposed on the core pathogenic process and that the nature of these derangements are important to symptoms and clinical presentation.
- That the core driver of the inability to sustain exertion seen in this disease is due to altered metabolic process(es), likely those concerning cellular respiration and the mitochondria.
I hope this study will lead to a much greater interest in the lipidome in ME and the differences between our cells’ respiration versus healthy patients. It will be important to determine if the metabolic dysfunction in ME is due to an intrinsic alteration of cellular respiration (for example, an acquired dysfunction in an mitochondrial enzyme) or are an extrinsic effect which functions to impair cellular respiration (for example, a loss of blood flow due to circulatory system dysfunction).
Lastly it should be noted that this study used the Canadian Consensus Criteria,9 and attempts to replicate it with inferior criteria such as Fukuda or SEID, such as some of our USA orgs are, inexplicably, fond of will likely produce widely divergent results.
Citations:
- Hoel F, Hoel A, Pettersen IK, Rekeland IG, Risa K, Alme K, Sørland K, Fosså A, Lien K, Herder I, Thürmer HL, Gotaas ME, Schäfer C, Berge RK, Sommerfelt K, Marti HP, Dahl O, Mella O, Fluge Ø, Tronstad KJ. A map of metabolic phenotypes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight. 2021 Aug 23;6(16):e149217. doi: 10.1172/jci.insight.149217. PMID: 34423789; PMCID: PMC8409979.
- Hokama Y, Uto GA, Palafox NA, Enlander D, Jordan E, Cocchetto A. Chronic phase lipids in sera of chronic fatigue syndrome (CFS), chronic ciguatera fish poisoning (CCFP), hepatitis B, and cancer with antigenic epitope resembling ciguatoxin, as assessed with MAb-CTX. J Clin Lab Anal. 2003;17(4):132-9. doi: 10.1002/jcla.10079. PMID: 12784262; PMCID: PMC6807878.
- Hokama Y, Empey-Campora C, Hara C, Higa N, Siu N, Lau R, Kuribayashi T, Yabusaki K. Acute phase phospholipids related to the cardiolipin of mitochondria in the sera of patients with chronic fatigue syndrome (CFS), chronic Ciguatera fish poisoning (CCFP), and other diseases attributed to chemicals, Gulf War, and marine toxins. J Clin Lab Anal. 2008;22(2):99-105. doi: 10.1002/jcla.20217. PMID: 18348309; PMCID: PMC6649096.
- Kennedy G, Spence VA, McLaren M, Hill A, Underwood C, Belch JJ. Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med. 2005 Sep 1;39(5):584-9. doi: 10.1016/j.freeradbiomed.2005.04.020. PMID: 16085177.
- Kennedy G, Spence V, Underwood C, Belch JJ. Increased neutrophil apoptosis in chronic fatigue syndrome. J Clin Pathol. 2004 Aug;57(8):891-3. doi: 10.1136/jcp.2003.015511. PMID: 15280416; PMCID: PMC1770396.
- Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, Onoe K, Fukuda S, Kawabe J, Takahashi K, Kataoka Y, Shiomi S, Yamaguti K, Inaba M, Kuratsune H, Watanabe Y. Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An ¹¹C-(R)-PK11195 PET Study. J Nucl Med. 2014 Jun;55(6):945-50. doi: 10.2967/jnumed.113.131045. Epub 2014 Mar 24. PMID: 24665088.
- Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, Onoe K, Fukuda S, Kawabe J, Takahashi K, Kataoka Y, Shiomi S, Yamaguti K, Inaba M, Kuratsune H, Watanabe Y. Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An ¹¹C-(R)-PK11195 PET Study. J Nucl Med. 2014 Jun;55(6):945-50. doi: 10.2967/jnumed.113.131045. Epub 2014 Mar 24. PMID: 24665088.
- Light AR, Bateman L, Jo D, Hughen RW, Vanhaitsma TA, White AT, Light KC. Gene expression alterations at baseline and following moderate exercise in patients with Chronic Fatigue Syndrome and Fibromyalgia Syndrome. J Intern Med. 2012 Jan;271(1):64-81. doi: 10.1111/j.1365-2796.2011.02405.x. Epub 2011 Jul 13. PMID: 21615807; PMCID: PMC3175315.
- Carruthers BM, Jain AK, De Meirleir KL, Peterson DL, Klimas NG, Lerner AM, Bested AC, Flor-Henry P, Joshi P, Powles ACP, Sherkey JA, van de Sande MI. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Clinical Working Case Definition, Diagnostic and Treatment Protocols. J Chronic Fatigue Syndrome. 2003 Jan;11(1)7-115. doi: 10.1300/J092v11n01_02 Epub 2011 Dec 04.
2 thoughts on “August and September Research Review – Part 1”